Evaporation Kinetics of Nano Water Droplets using Coarse-Grained Molecular Dynamic Simulations
I worked as a research assistant in the Environmental Process Modeling Centre at
Nanyang Environmental and Water Research Institute in Singapore between June-August 2018 and May-July 2019.
One of my primary research focus was on some of the science behind interfacial water evaporation.
My research was only possible with my fortunate opportunity to use the Nanyang Technological University's
High Performance Computing Centre to run large scale simulations using
LAMMPS .

Molecular dynamic simulations are the basis of the Folding@Home project, which directly models every single atomic interactions of a protein biomolecule. By simulating a nanoscale system, we can take meaningful statistical averages and interpret the phenomena in macroscopic terms. Though I am not qualified to explain why the data from this type of numerical modeling has any validity in real life, this paper may provide good insight on this topic.
Read Evaporation Kinetics of Nano Water Droplets using Coarse-Grained Molecular Dynamic Simulations here.

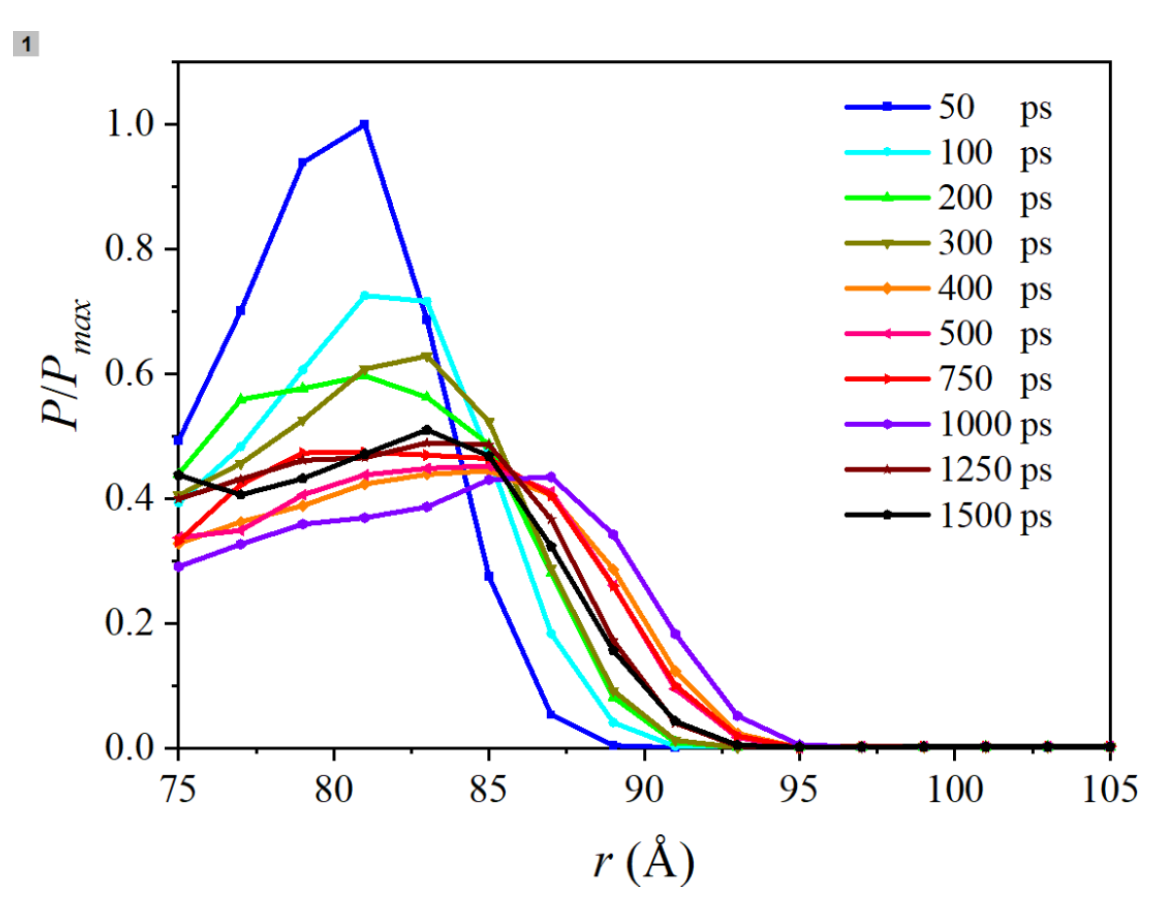
Read Evaporation Kinetics of Nano Water Droplets using Coarse-Grained Molecular Dynamic Simulations here.